Research

Mechanosensitive Ion Channels
The mechanosensitive PIEZO ion channels are implicated in many clinical conditions, including lymphedema, xerocytosis, inflammation, chronic pain, and cancer. However, the molecular mechanisms by which PIEZO senses force remain largely unknown. Using molecular dynamics (MD) simulations, we identified an agonist binding site on the transmembrane domain of PIEZO1 (Nat.Commun.2019), generated the first PIEZO1 open-state model by modulating membrane curvature (Commun.Biol.2020), and quantified how anionic lipids such as PIP2 and PS modulate the mechanical properties of asymmetric membrane bilayer (J.Chem.Phys.2021). During these MD studies, we also found that extra caution is required during the initial MD equilibrium to avoid overestimating lipid density in the channel pore (Biophys.Rep.2022). Additionally, we discovered new PIEZO1 agonists using virtual screening and free energy approaches (PNAS2023).
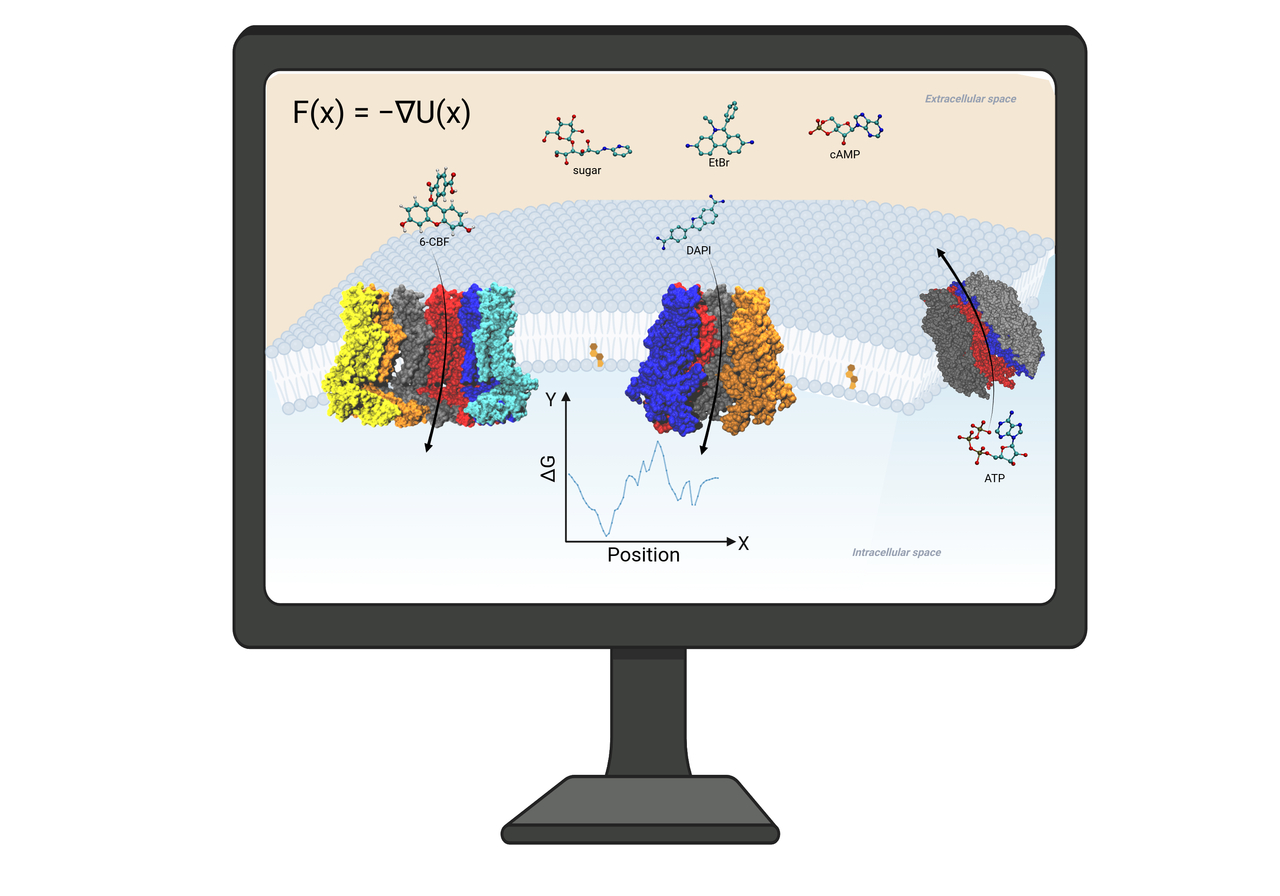
Large-pore Channel Permeability and Selectivity
Eukaryotic large-pore channels are permeable to biological signaling molecules. To understand how these large pores selectively permeant molecules, we employed Hamiltonian replica-exchange umbrella sampling, Markovian milestoning, and voltage-driven flux MD simulations to investigate the free energy and kinetics of dye molecules and cAMP permeation through connexin hemichannels (Biophys.J.2016 & 2021; PNAS2024). As a benchmark, we compared and validated our MD approaches in predicting the single-channel permeability of a carbon nanotube (Front.Mol.Biosci.2022). Statistical analysis of MD trajectories has also been useful for understanding the gating mechanism and disease mutations in connexin and gap junctional channels (2016; Am.J.Physiol.2018; J.Gen.Physiol.2019). We summarized these findings and methods in JGP.2024.
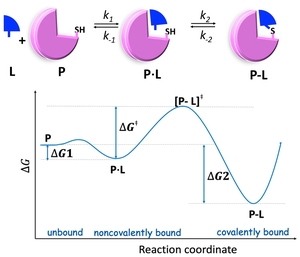
Binding Free Energy of Covalent Drug
Predicting covalent binding affinity typically requires quantum mechanical (QM) calculations, which are prohibitively expensive for drug discovery. We developed a binding free energy simulation framework that estimates reversible covalent binding affinity without relying on costly QM calculations (JACS 2017; JCIM2019; viewpoint JCIM2021). This computational approach has enabled our design of calpain-2 selective inhibitors for neuroprotection follwing brain injury (Neurotherapeutics2023; Pharm.Res.2024).
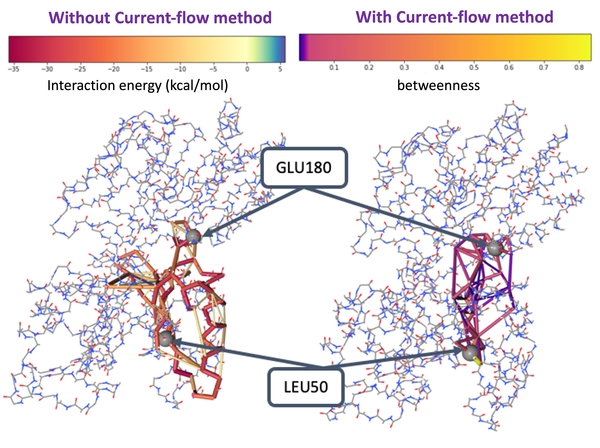
Robust Protein Allosteric Network Analysis
Using current-flow betweenness, we show that protein dynamic signaling propagates between amino acids can be mapped out similarly to the electric current flow using Kirchhoff’s law. Our method provides a more robust network topology compared to the commonly used correlation or interaction networks (JCTC2019, Methods Mol.Biol.2021). Applying this method, we identified an allosteric pathway between the kinase regulatory domain and the catalytic domain that disease-causing mutations can bypass, leading to abnormal bone formation signaling (Plos Comput.Biol.2017; J. Comput. Chem 2019). We are now developing a user-friendly website called “Current-Flow Allostery”.
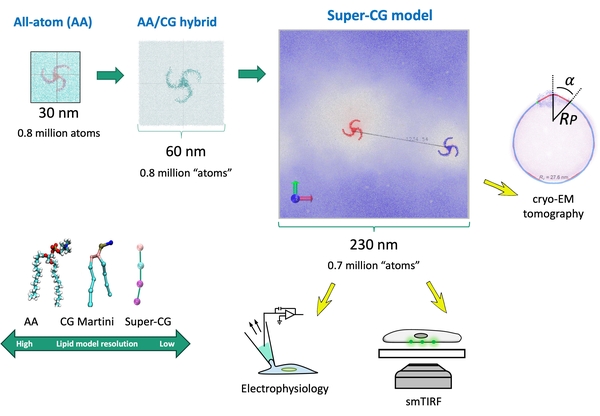
Hybrid and Coarse-Grained Membrane Protein Simulations
Due to the large size of many membrane proteins, simulating their gating motions, protein-lipid, and protein-protein interactions using atomistic MD is challenging. This is especially true for ion channels that deform membranes beyond tens of nanometers (e.g., PIEZOs). To address the sampling issue arising from the size of membrane systems, we employed a hybrid-resolution MD approach (JCTC2024) to study the force-from-lipid gating motions and a solvent-free coarse-grained model (bioRxiv) to investigate channel clustering in membrane with submicron dimensions.
Here are two videos that provide a visual representation of the PIEZO2 clockwork gating mechanism we uncovered using our hybrid-resolution MD approach:
- Global PIEZO2 motion: https://www.youtube.com/watch?v=wcMu-1KSRlw
- Pore clockwork motion: https://www.youtube.com/watch?v=NIlxsekgcrM